Glycogen Metabolism and Glycogen storage Diseases
GLYCOGEN:
Glycogen is the major storage carbohydrate in animals, corresponding to starch in plants; it is a branched polymer of α-D-glucose. It is stored mainly in liver and muscle. Due to more muscle mass, the quantity of glycogen in muscle (250gm) is about 3 times higher than that in the liver (75 gm).
Functions:
- Liver glycogen helps to maintain blood glucose level: Liver glycogen functions to store and export glucose to maintain blood glucose level between meals. After 12-18 hrs of fasting, liver glycogen is almost totally depleted.
- Muscle glycogen serves as a fuel reserve for the supply of ATP during muscle contraction.
- Although muscle glycogen doesn’t yield free glucose (as muscle lacks Glucose-6-phosphatase), pyruvate formed by glycolysis in muscle can undergo transamination to alanine, which is exported from muscle and used for gluconeogenesis in liver.
Structure:
- homopolysaccharide composed of α-D-glucose
- glucose units are held together by α-1,4 glycosidic linkages
- α-1,6 glycosidic linkages at branching
GLYCOGEN METABOLISM:

a. Glycogenesis: The synthesis of glycogen from glucose occuring in the cytosol in the presence of ATP, UTP and glucose is called glycogenesis.
Steps:
- Glucose is converted into glucose-6-phosphate by the action of glucokinase or hexokinase.
- Glucose-6-phosphate is converted into glucose-1-phosphate by the action of Phosphoglucomutase, passing through an obligatory intermediate step of glucose-1,6-bisphosphate.
- Glucose-1-phosphate is converted into UDP-glucose by the action of Uridyl Transferase (also called UDP-glucose pyrophosphorylase) and pyrophosphate is formed, which is hydrolyzed by pyrophosphatase into 2 molecules of Pi.
- Glucose molecules are assembled in a chain by glycogen synthase, which must act on a pre-existing glycogen primer or glycogenin (small protein that forms the primer). The mechanism for joining glucose units is that glycogen synthase binds to UDPG, causing it to break down into an oxonium ion, also formed in glycogenolysis. This oxonium ion can readily add to the 4-hydroxyl group of a glucosyl residue on the 4 end of the glycogen chain.
- Branches are made by branching enzyme (also known as amylo-α(1:4)->α(1:6)transglycosylase), which transfers the end of the chain onto an earlier part via α-1:6 glucosidic bond, forming branches, which further grow by addition of more α-1:4 glucosidic units.
Overall reaction:
(Glucose)n + Glucose + 2ATP = (Glucose)n+1 + 2ATP + Pi
b. Glycogenolysis: The degradation of stored glycogen in liver and muscle constitutes glyogenolysis. It is not the reverse of glycogenesis but is a separate pathway.
Steps:
1. First step:
The overall reaction for the 1st step is:
Glycogen (n residues) + Pi = Glycogen (n-1 residues)+ G1P
Here, glycogen phosphorylase cleaves the bond at the 1 position by substitution of a phosphoryl group. It breaks down glucose polymer at α-1-4 linkages until 4 linked glucoses are left on the branch. Glycogen phosphorylase can be used as a marker enzyme to determine glycogen breakdown.
2. Second step:
The 2nd step involves the debranching enzyme that moves three remaining glucose units to another 1,4 end of glycogen. The final action of the debranching enzyme is the hydrolysis of the glucose attached as a 1,6-branched mono residue, giving one free glucose molecule. This is the only case in which a glycogen metabolite is not glucose-1-phosphate.
3. Third step:
The 3rd and last stage converts G1P (glucose-1-phosphate) to G6P (glucose-6-phosphate) through the enzyme phosphoglucomutase.
Degradation of glycogen by lysosomal acid maltase:
- Acid maltase or alpha-1,4 glucosidase is a lysosomal enzyme that continuously degrades a small quantity of glycogen.
- Deficiecy results in glycogen accumulation causing Pompe’s disease.
Mechanisms of regulation of glycogenesis and glycogenolysis:
- Allosteric mechanism
- Hormonal mechanism
- Influence of calcium
1. Allosteric regulation:
- The control is carried out in such a way that glycogen synthesis is increased when substrate and energy levels are high. On the other hand, glycogen breakdown is enhanced when glucose concentration and energy levels are low.
2. Hormonal regulation:

- The hormones, through a complex series of reactions bring, about covalent modification : phosphorylation and dephosphorylation of enzyme proteins.
- cAMP acts as a second messenger for hormones.
3. Influence of calcium:
- Calcium ions are released from sarcoplasmic reticulum during muscle contraction
- Calcium binds to calmodulin (calcium modulating protein) and directly activates phosphorylase kinase without the involvement of cAPM-dependent protein kinase.
- Muscle phosphorylase kinase activates glycogen phosphorylase.
Increased glucagon or epinephrine level = increased glycogenolysis
Increased insulin = increased glycogenesis
GLYCOGEN STORAGE DISEASES (GSD):
The metabolic defect concerned with the glycogen synthesis and degradation are collectively referred to as glycogen storage diseases. Glycogen storage diseases are inherited.
Common Types:
Mnemonics: VP CAM HT
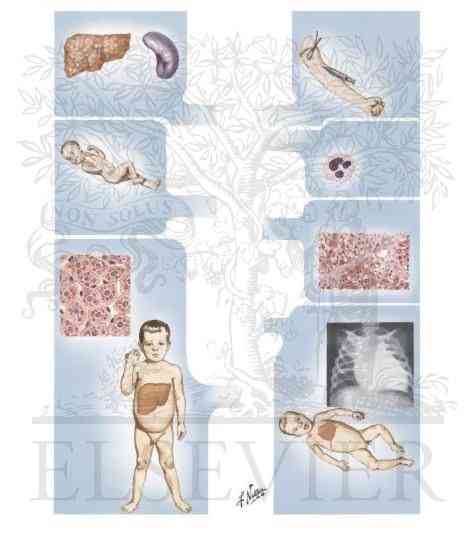
Type I: von Gierke’s disease
- Enzyme defect: Glucose-6-phosphate
- Organs involved: Liver, kidney and intestine
- Features: Glycogen accumulation in liver and renal tubule cells; enlarged liver and kidney; hypoglycemia; lactic acidemia; ketosis, hyperlipemia; gouty arthritis
Type II: Pompe’s disease
- Enzyme defect: Lysosomal α-1,4 glucosidase (acid maltase)
- Organs involved: All organs
- Features: Glycogen accumulation in lysosomes; juvenile onset variant: muscle hypotonia, death from heart failure by age 2; adult onset variant: muscle dystrophy; enlarged liver and nervous system is also affected.
Type III: Cori’s disease or limit dextrinosis
- Enzyme defect: Amylo α-1,6 glucosidase (debranching enzyme)
- Organs involved: liver, muscle. heart, leukocytes
- Features: Accumulation of branched chain glycogen; liver enlarged; hypoglycemia; muscle weakness.
Type IV: Andersen’s disease or amylopectinosis
- Enzyme defect: Glucosyl 4-6 transferase (branching enzyme)
- Organs involved: Most tissues
- Features: Hepatosplenomegaly; liver cirrhosis; accumulation of glycogen with few branches; death from heart or liver failure by age 5.
Type V: McArdle’s syndrome
- Enzyme defect: Muscle glycogen phosphorylase
- Organs involved: Skelteal muscle
- Features: Muscle glycogen stores very high; poor exercise tolerance; muscle cramps; blood lactate and pyruvate very low after exercise.
Type VI: Her’s disease
- Enzyme defect: Liver glycogen phosphorylase
- Organs involved: Liver
- Features: Hepatomegaly; accumulation of glycogen in liver; mild hypoglycemia; ketosis.
Type VII: Tauri’s disease
- Enzyme defect: Muscle and erythrocyte phosphofructokinase 1
- Organs involved: Skeletal muscle, erythrocyte
- Features: Muscle cramps due to exercise; blood lactate not elevated; hemolytic anemia; poor exercise tolerance.
Sources: Harper’s Biochemistry, Satyanarayan’s Biochemistry, Wikipedia

5 Comments
Increased glucagon or epinephrine level = increased glycogenolysis
Increased insulin = increased glycogenolysis
should be:
Increased glucagon or epinephrine level = increased glycogenolysis
Increased insulin = increased glycogenesis
Thanks for the correction Ronald.
This brings back memories (or nightmares) of Chemistry 1 & 2 in college.
Nice class note. I could print it for my reading.
It is more like a lecture notes for quick reciew of the topic.
Comments are closed.