Yellowish discoloration of eyes and body for 15 days
Excessive crying for 2 days
Decrease responsiveness for 1 day
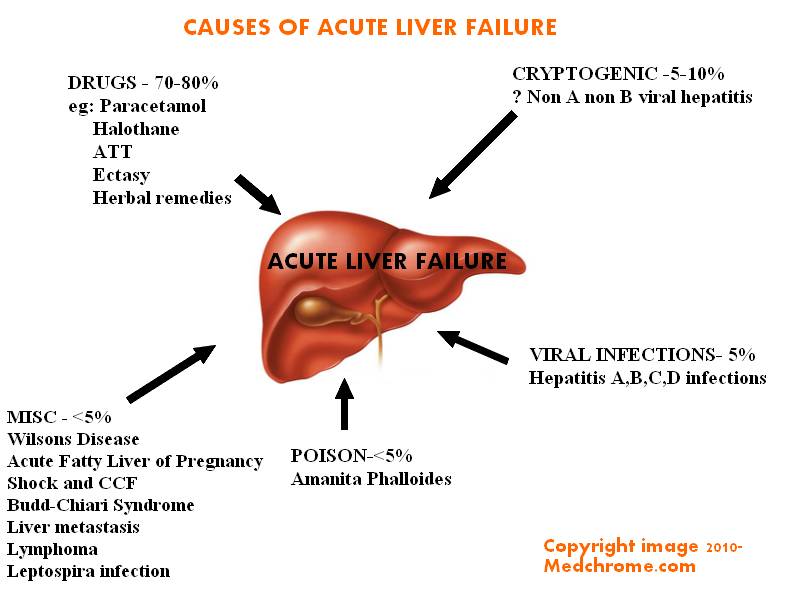
Causes of acute liver failure In Adults
HOPI
As per mother, the child was apparently well before, developed yellowish discoloration, beginning in the eyes and progressed over whole body over next 15 days associated with passing whitish colored stool over 1 week, diarrhea -, bulky stool -, reddish or black staining of stool –,bluish patches-, Petechia -, bleeding – ,No h/o abdominal distension
feeding well and had normal activity until 2 days back, onset of shrill cry and excessive irritability for 2 days with vomiting 1 episode, non-bilious, not blood tinged, containing food particles. Then child developed decreased responsiveness , increased drowsiness. Stopped feeding.
Pallor noticed by parents +
No abnormal body movements, no vacant stare and unrolling of eyes
No fever
No bleeding manifestations
Fast breathing was present since 2 days however there was no chest in drawing, cough or noisy breathing
Child was passing urine normally
Stool was not passed since 2 days
No h/o swelling of limbs or joints, No h/o swelling at injection sites
No h/o jaundice or bleeding manifestations in the past, No h/o blood transfusions
No h/o exposure to any drug, or toxins
No floppiness or stiffness of body
No h/o recurrent chest infections
No h/o weight loss
Child was born via Normal vaginal delivery at KMCTH, was AGA (2.5 kg) term baby
Had NNJ for which he was admitted and phototherapy was given for 2 days. Resolved and discharged.
Immunized as per EPI schedule till 14 weeks
No h/o consanguinity
No family h/o jaundice, liver disease or chronic illness
Lives in joint family
Only child of parents
On h/o fetal loss or infant deaths.
Developmental history
Motor- neck holding/ rolling over ~ 5 months
Smiles- 3 months
Cooing- 3months
Hearing/Vision- normal as per parents
Historical diagnosis
Hepatic encephalopathy
Sepsis
Intracranial bleeding – late onset hemorrhagic disease
Hemolytic anemia
On examination at ER
Child was unconscious GCS=8/15, tachypneic
AF- bulged
No facial dysmorphism
Ecchymosis at cannulation site, with swelling.
Pallor +++ icterus + Edema –
Vitals-
–Temp-97F
–RR= 60/min SCR-
–BP=60/30 mmHg
–HR-150/min
–SpO2= 86% with out O2
Wt-7.5 kg- Normal for age
Per Abdominal Examination-
–Soft, non-tender, distension-
–No venous prominence or scar marks
–Fluid thrill-, SD-
–Liver + 5 cm below SCM at MCL, firm, smooth and Spleen – not palpable, BS+
CNS-
-GCS-8/15 ( E2V1M5)
–Pupils-B/L dilated 5/6 mm, sluggish
–Tone- Increased B/L
–Reflex- brisk knee jerk, clonus –
–Plantars upgoing b/l
–AF – bulged
Chest/CVS- Normal
Hematoma + over thigh at cannulation site, ecchymosis over the area. No Petechia/purpura over body and mucus membranes.
No joint swelling or swelling over muscles
Summary
5 months Male child developmentally normal born of non-consanguinous marriage was admitted with complains of yellowish discoloration of skin and sclera for 15 days, associated with white stools for 1 week followed by excessive crying and vomiting for 2 days and decrease responsiveness for 1 day. No fever, seizure, bleeding manifestations, exposure to drugs or toxins, jaundice or blood transfusion in the past.
No h/o sibling death or fetal loss.
On examination- child was unconsious, severely pale, icterus and was in shock, ecchymosis, hematoma + rt thigh
Liver was palpable 5 cm, firm, spleen-
GCS-8/15, AF was bulged with b/l sluggish reacting pupils, increased tone in LL and exaggerated DTR.
Diagnosis
Provisional –Fulminant Hepatic Failure
Differential-
Septic Shock
Late Hemorrhagic disease ( Vit K deficiency)
Hemolytic anemia ( Autoimmune )
Management at Pediatric ER
Oxygen via head box
Fluid bolus- 20ml/kg – twice
Dopamine @ 10 mic/kg/min
IVF maintenance
Inj Vit K
IV Antibiotic- Cefotaxime + Amikacin
Blood arranged
Received II FFP and I whole blood
Inj Phenytoin maintenance
Inj Mannitol Loading and maintenance
Syr Lactulose
NAC started
Admitted at Ped Ward with High Risk consent
CAUSE OF FULMINANT HEPATIC FAILURE AT INFANCY IS TO BE SOUGHT FOR
Investigation reports
Hb-2.1 g% PCV-6.4
TC-17900, 49N, 44L Plateletes- 340,000
RBS- 96mg/dl
Urea- 85 mg/dl Creatinine- 0.96 mg/dl
Na+- 147 K+- 4.4
Total bilirubin- 2.3 mg/dl direct-1.7 mg/dl
SGPT- 87, SGOT-124, ALP-257, Albumin-4.5g/dl
Calcium- 2.4
PT- 20 INR-1.5 , PT/INR- INR -9.0 ( outside)
aPTT- normal
CT scan- SDH Rt. FPTO, Rt PCA infarcts
2nd DOA
Operative procedure- Rt FTP Craniotomy with Rt Subtemporal craniectomy with evacuation of SDH with loose duroplasty
Findings- Rt FTP acute SDH, no active bleeding source, Clot overlying brain was removed.
Urine-Albumin +++ Reducing substance +
Sugar/rbc- nil
ABG- pH-7.52 pCO2-22 pO2-132 HCO3-19 Lac-1.4
PO4- 7.8
Alfa Feto Protein= 26.1 ( ref <7.51)
HBsAg /HAV/HEV- NR
Serum Ferritin- Normal Serum Iron-59 microg/dl ( Normal)- Neonatal hemochromatosis
Urine +ve for reducing sugarà?Galactosemia, Fanconi synd
Eye evaluation- No f/o cataract or chorioretinitis
USG abdomen- Hepatomegaly, GB calculi 7 mm, Dilated CBD 5 mm calculus in lower end, Thick irregular UB s/o cystitis
Urine Metabolic screening- weakly +ve FeCl3 test, AAs –ve, non-specific reducing sugar +, FA screen not done – Fanconi Syndrome less likely
Discharged with Dx-
–Fulminant Hepatic failure (? Metabolic) with S/P rt craniectomy with evacuation of SDH.
Adviced for follow with TMS reports.
J Comput Assist Tomogr. 2002 Jan-Feb;26(1):69-72. Is coagulopathic liver disease a factor in spontaneous cerebral hemorrhage? Lee HJ, Hinrichs CR.
Hepatic dysfunction is a common cause of thrombocytopenia and coagulopathy and has been reported as a causal factor in spontaneous intracranial hemorrhage. We attempt to define the prevalence of intracranial hemorrhage in patients with severe liver disease and coagulopathy.
Contrary to past reports, we find no instance of spontaneous intracranial hemorrhage in patients with coagulopathic liver disease presenting with acute mental status changes. Therefore, unless associated with trauma, spontaneous intracranial hemorrhage in coagulopathic liver disease is very uncommon.
Fulminant Hepatic failure
IAP consensus -Pediatric ALF definition as
a) evidence of liver dysfunction within 8 weeks of onset of symptoms (neonates may have only deranged liver functions without overt symptoms)
(b) uncorrectable (6-8 hours after administration of one dose of parenteral vitamin K) coagulopathy with International Normalized Ratio (INR) >1.5 in patients with hepatic encephalopathy, or INR> 2.0 in patients without encephalopathy and
(c) No evidence of chronic liver disease either at presentation or in the past
Staging of encephalopathy
in infants and children is difficult as compared to adults.
Grades I and II are indistinguishable with clinical features of inconsolable crying, inattention to task: with normal or exaggerated deep tendon reflexes.
Grade III encephalopathy manifests as somnolence, stupor, combativeness and hyperreflexia.
In grade IV, child is comatose [arousable with painful stimuli (IVa) or no response (IVb)] with absent reflexes and decerebration or decortication.
Acute Liver Failure in Children: The First 348 Patients in The Pediatric Acute Liver Failure Study Group
J Ped, 2006
The etiology of ALF in 348 children from birth to 18 years with ALF were included
Five studies published between 1996 and 2007 studies from India (Chandigarh, Vellore, Delhi, Kolkata and Pune) enrolling 215 children
Acute viral hepatitis to be the commonest cause, either alone or in combination (overall 61-95%: hepatitis A 10-54%; hepatitis E 3-27% ; hepatitis B 8-17%; and multiple viruses 11-30%- commonest being hepatitis A+E).
Drugs were responsible for ALF in 6-8% cases and other causes in 9-10.5%.
Etiology remained unestablished in 6-22% patients.
There are no published data from India on ALF in neonates and infants.
Fulminant Hepatic failure- Causes in Infants and children
Tyrosinemia Type 1
Typical age-2 -6 months (rarely <1 mth and >1 yr of life).
Defect -fumarylacetoacetate hydrolase (FAH)
Coagulopathy with or without cholestatic jaundice, hypoglycemia, hepatomegaly, ascites
An acute hepatic crisis commonly heralds the onset of the disease – intercurrent illness
Fever, irritability, vomiting, hemorrhage, hepatomegaly, jaundice, elevated levels of serum transaminases, and hypoglycemia
An odor resembling boiled cabbage may be present ( methionine metabolites )
Most hepatic crises resolve spontaneously, but may progress to liver failure and death.
Between the crises, varying degrees of failure to thrive, hepatomegaly, and coagulation abnormalities often persist. Cirrhosis and eventually hepatocellular carcinoma occur with increasing age. Carcinoma is unusual before 2 yr of age.
Episodes of acute peripheral neuropathy resembling acute porphyria occur in ≈40% of affected children.
Crises typically last 1 to 7 days but recuperation from paralytic crises can be prolonged.
Renal involvement is manifested as a Fanconi-like syndrome with normal anion gap metabolic acidosis, hyperphosphaturia, hypophosphatemia, and vitamin D–resistant rickets. Nephromegaly and nephrocalcinosis may be present on ultrasound examination.
Hypertrophic cardiomyopathy and hyperinsulinism are seen in some infants
Laboratory Findings
•The presence of elevated levels of succinylacetone in serum and urine is diagnostic for tyrosinemia type I
• In untreated patients, α-Fetoprotein level is increased, often markedly
•Liver-synthesized coagulation factors are decreased in most patients; serum levels of transaminases are often increased, with marked increases being possible during acute hepatic episodes.
•Serum concentration of bilirubin is usually normal but can be increased with liver failure.
Treatment and Outcome
•A diet low in phenylalanine and tyrosine can slow but does not halt the progression of the condition.
•The treatment of choice is nitisinone (NTBC), which inhibits tyrosine degradation at 4-HPPD, prevents acute hepatic and neurologic crises.
Although nitisinone stops or greatly slows disease progression, some pretreatment liver damage is not reversible.
Therefore, patients must be followed for development of cirrhosis or hepatocellular carcinoma.
On imaging, the presence of even a single liver nodule usually indicates underlying cirrhosis. Most liver nodules in tyrosinemic patients are benign but current imaging techniques do not accurately distinguish all malignant nodules.
Liver transplantation is an effective therapy for tyrosinemia type I and alleviates the risk of hepatocellular carcinoma.
Course
•The earlier the presentation, the poorer is the prognosis.
•The 1 yr mortality, which is about 60% in infants who develop symptoms before 2 mo of age, decreases to 4% in infants who become symptomatic after 6 mo of age.
Tyrosinemia type 1 should be suspected in infants with severe coagulopathy even in the absence of other signs of liver failure. Pediatrics. 1999 Mar;103(3):675-8
A Review of 10 Cases from the University of Pittsburgh 1990
The outcome of 10 patients with tyrosinemia, who underwent orthotopic liver transplantation at the University of Pittsburgh, was reviewed.
The indications for transplantation were:
-hepatoma in three,
-acute liver failure in two, and
-progressive chronic liver disease in five.
Seven patients alive 6 months to 6½ years following transplantation. Of these seven patients, six have normal liver function and a good performance status. One is awaiting retransplantation for chronic rejection. Hepatocellular carcinoma (HCC) was found either preoperatively or incidentally in five patients, all older than 2 years at the time of their transplant. Four of these are alive and well without evidence of tumor with follow-ups between 3½ and 6½.
This experience suggests that liver transplantation should be considered seriously for children with hereditary tyrosinemia who are more than 2 years of age because beyond that age the incidence of hepatocellular carcinoma (HCC) increases substantially.
Mitochondrial cytopathy
Onset in the first week of life or later, transient hypoglycemia, neurological involvement in form of severe hypotonia, myoclonus or psychomotor retardation.
Diagnosis:
–Plasma lactate >2.5 mmol/L, molar ratio of plasma lactate/pyruvate > 20:1, paradoxical increase in plasma ketone bodies or lactate after meals
–Urinary analysis by mass spectroscopy
–Genetic mutational analysis for respiratory chain disorders and tandem mass spectrometry for fatty acid oxidation defects.
Galactosemia
Typical Age- Classically by 2nd half of week of birth after starting of milk feeding.
Feeding intolerance, vomiting, diarrhea, jaundice, hepatomegaly, lethargy and hypotonia after milk feeding is started; hypoglycaemia, sepsis (particularly E.coli), cataract and developmental delay.
Diagnosis: Urine positive for non-glucose reducing substances while on lactose feeds; confirmation by blood Galactose-1 phosphatase uridyl transferase enzyme assay.
Presents usually few hours to sometimes weeks after birth as hypoglycemia, coagulopathy, jaundice, anemia, ascites, anasarca, and splenomegaly with shrunken liver.
Diagnosis: Transaminases- N or low, hypoalbuminemia, hypofibrinogenemia, thrombocytopenia, high serum ferritin, low serum transferrin, high transferrin saturation.Lip or salivary gland biopsy à iron deposition; MRI pancreas àlow signal intensity on T2 imaging.
Treatment: Anti-oxidants (acetyl-cysteine and Vitamin E), high dose IVIG in combination with exchange transfusion; liver
Herpes simplex infection
No positive history in 60 % to 80 % of mothers. Suspect in a sick neonate presenting in first week of life especially if bacterial cultures are not growing anything. Search for vesicles, particularly on scalp.
Diagnosis: Viral cultures form vesicles, oropharynx, conjunctiva, blood or CSF; PCR diagnosis from blood or CSF.
Treatment: High dose (60 mg/kg/d) acyclovir for 21 days or till PCR is negative. Necessary to document negative CSF-PCR at end of therapy.
Neonatal Intrahepatic Cholestasis (Citrullinemia Type II-Neonatal Form)
Age at presentation- usually start before 1 yr of age
Cholestatic jaundice with mild to moderate direct hyperbilirubinemia
marked hypoproteinemia
Clotting dysfunction (increased prothrombin time and partial thromboplastin times), and
increased serum γ-glutamyltranspeptidase (GGTP) and alkaline phosphatase activities; liver transaminases are usually normal.
Plasma concentrations of ammonia and citrulline are usually normal, but moderate elevations are reported.
There may be increases in plasma concentrations of methionine, tyrosine, alanine, and threonine. Elevated levels of serum galactose may occur, but all enzymes involved in galactose metabolism are normal. The reason for hypergalactosemia is not known.
Increased serum α-fetoprotein is also present. (resembles tyrosinemia type I ,urinary excretion of succinylacetone –ve).
Liver biopsy shows fatty infiltration, cholestasis with dilated canaliculi, and a moderate degree of fibrosis.
Self-limiting -majority of infants recover spontaneously by 1 yr of age with only supportive and symptomatic treatment.
Treated with low-protein diet
Hepatic failure requiring liver transplantation has occurred in a few cases. The diagnosis should be considered in cases of unexplained neonatal epatitis with cholestasis.
Data on the long-term prognosis and the natural history of the condition are limited.
Infectious Causes
HBV is also responsible for some cases of fulminant liver failure in the absence of serologic markers of HBV infection but with HBV DNA found in the liver. Hepatitis C and E viruses are uncommon causes of fulminant hepatic failure in the United States. Patients with chronic HCV are at risk if they have superinfection with HAV. Epstein-Barr virus, herpes simplex virus (HSV), adenovirus, enteroviruses, cytomegalovirus, parvovirus B19, human herpesvirus-6, and varicella-zoster infections can also produce fulminant hepatitis in children.
Fulminant hepatic failure can also be caused by autoimmune hepatitis in ~5% of cases. Patients have a positive autoimmune marker (e.g., antinuclear antibody, anti–smooth muscle antibody, liver-kidney microsomal antibody, or soluble liver antigen) and possibly an elevated serum IgG level. Liver histology, if a biopsy can be safely done, might support the diagnosis.
Acute liver failure is a common feature of hemophagocytic lymphohistocytosis (HLH) caused by several gene defects, infections by mostly viruses of the herpes group, and a variety of other conditions including organ transplantation and malignancies. Impaired function of natural killer (NK) cells and cytotoxic T-cells (CTL) with uncontrolled hemophagocytosis and cytokine overproduction is characteristic for genetic and acquired forms of HLH. Biochemical markers include elevated ferritin and triglycerides and low fibrinogen.
An idiopathic form of fulminant hepatic failure
Accounts for 40-50% of cases in children, 15% in Infants
Various hepatotoxic drugs and chemicals can also cause fulminant hepatic failure. Predictable liver injury can occur after exposure to carbon tetrachloride or Amanita phalloides mushroom or after acetaminophen overdose.
Acetaminophen, halothane, isoniazid, or sodium valproate.
Ischemia and hypoxia resulting from hepatic vascular occlusion, severe heart failure, cyanotic congenital heart disease, or circulatory shock can produce liver failure.
Intensive Care of ALF
Cerebral Edema/Intracranial Hypertension
Grade I/II Encephalopathy
Consider transfer to liver transplant facility and listing for transplantation
Brain CT: rule out other causes of decreased mental status; little utility to identify cerebral edema
Avoid stimulation, avoid sedation if possible
Antibiotics: surveillance and treatment of infection required; prophylaxis possibly helpful
Lactulose: possibly helpful
Grade III/IV Encephalopathy
Continue management as for Grade I/II HE +
Intubate trachea (may require sedation)
Elevate head of bed
Consider placement of ICP monitoring device
Immediate treatment of seizures required; prophylaxis of unclear value
Mannitol: use for severe elevation of ICP or first clinical signs of herniation
Hyperventilation: effects short-lived; may use for impending herniation
Infection
Surveillance for and prompt antimicrobial treatment of infection required
Antibiotic prophylaxis possibly helpful but not proven
Coagulopathy
Vitamin K: give at least one dose
FFP: give only for invasive procedures or active bleeding
Platelets: give for platelet counts 10,000/mm3 or invasive procedures
Recombinant activated factor VII: possibly effective for invasive procedures
Prophylaxis for stress ulceration: give H2 blocker or PPI
Hemodynamics/Renal Failure
Pulmonary artery catheterization
Volume replacement
Pressor support (dopamine, epinephrine, norepinephrine) as needed to maintain adequate mean arterial pressure
Avoid nephrotoxic agents
Continuous modes of hemodialysis if needed
NAC, prostacyclin: effectiveness unknown
Vasopressin: not helpful in ALF; potentially harmful.
Consider nutrition: enteral feedings if possible or total parenteral nutrition
Indicators of Poor Prognosis in Patients With ALF
Etiology
1.Idiosyncratic drug injury
2.Acute hepatitis B (and other non-hepatitis A viral infections)
3.Autoimmune hepatitis
4.Mushroom poisoning
5.Wilson disease
6.Budd-Chiari syndrome
7.Indeterminate cause
Coma grade on admission-III/IV
Prognostic – King’s College Criteria:
Summary
In 40-60% cases of fulminant hepatic failure, cause are indeterminate in children and in 15% cases in infants.
Metabolic Diseases are the most common cause of FLF in infancy in contrast to Acetaminophen toxicity and infective causes in older age groups( >3 yrs).
Reaching to diagnosis of Metabolic cause of FLF is hindered by unavailability of sophisticated test and cost factors.
Routine metabolic screening for neonates not available in our settings- can pick disease early.
Liver Biopsy is an important investigation when etiology becomes elusive
ICU care and close monitoring is needed as patient can have rapid deterioration.
OLT might be the only life saving treatment in many cases, patients who develop FHF who undergo transplantation have worse outcome compared to those with other indications like CLD.
OLT is not possible in Nepal. Nearest center is India- Apollo Hospital where children OLT has been done with great success rates.
References-
1.Nelson Textbook of Pediatrics 19 th Edition
2.The Management of Acute Liver Failure Julie Polson and William M. Lee- AASLD
3.Management of Acute Liver Failure in Infants and Children: Consensus Statement of the Pediatric Gastroenterology Chapter, Indian Academy of Pediatrics
4.Acute Liver Failure in Children Joel B. Cochran, DO and Joseph D. Losek, MD, Pediatric Emergency Care Volume 23, Number 2, February 2007
5.Acute Liver Failure in Children: The First 348 Patients in The Pediatric Acute Liver Failure Study Group- J Pedia 2006