Congenital Adrenal Hyperplasia or CAH
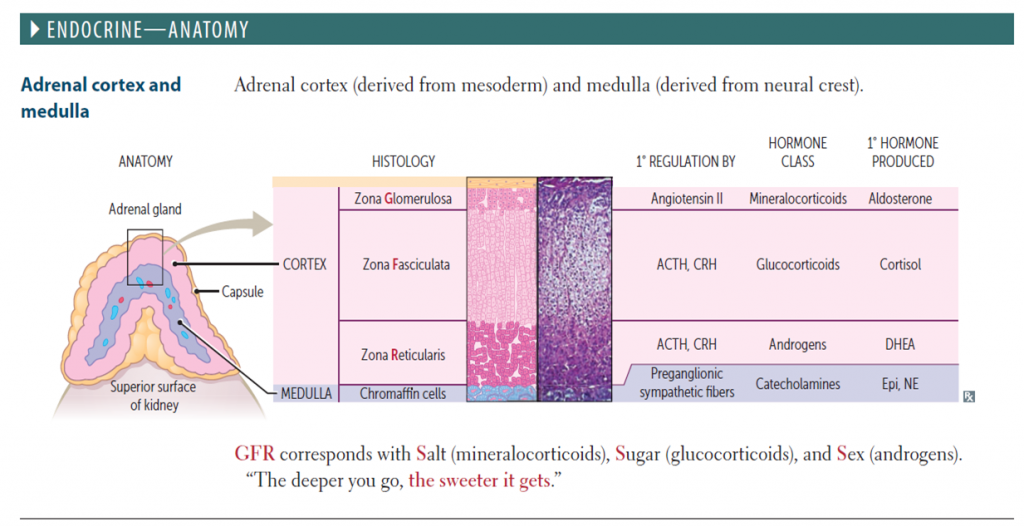
Congenital adrenal hyperplasia (CAH) is a family of autosomal recessive ( AR ) disorders of cortisol biosynthesis. Cortisol deficiency increases secretion of corticotropin (adrenocorticotropic hormone [ACTH]), which, in turn, leads to adrenocortical hyperplasia and overproduction of intermediate metabolites.
Epidemiology: The incidence of classical CAH in general population is between 1 in 13000 to 1 in 15000 live births. In a small study done in India, the incidence was found to be 1:2575. However large national survey has not been done in India. In a study done in India between 2007 to 2013, 11200 neonates were included out of which 15 had high 17-OHP ( 17 Hydroxyprogesterone) and 4 were confirmed to be CAH by repeat 17OHP along with a short Synacten test.
More than 90% of the cases of CAH is due to 21-hydroxylase deficiency . Approximately 70% of affected infants have the salt losing form and 30% have the simple virilizing form of the disorder.
In the United States, CAH is less common in African Americans compared with white children (1 : 42,000 vs 1 : 15,500). Nonclassic disease has a prevalence of approximately 1 in 1,000 in the general population but occurs more frequently in specific ethnic groups such as Ashkenazi Jews and Hispanics.
Pathophysiology and Clinical Variation:
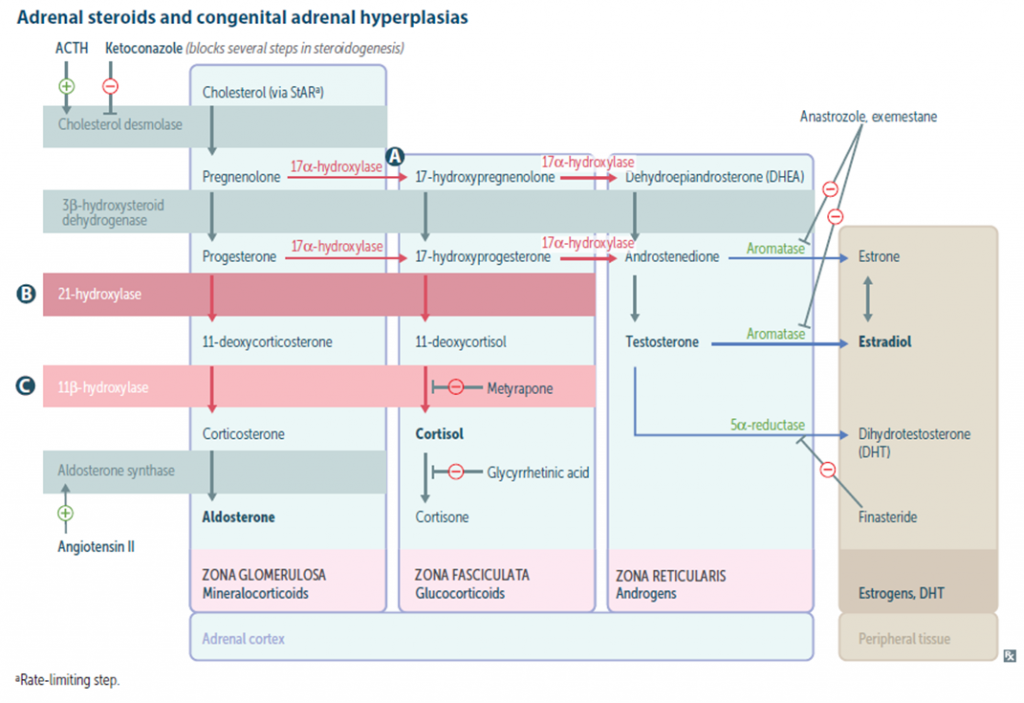

Classification:
- 21-Hydroxylase deficiency
- 11β-Hydroxylase deficiency
- 3β-HSD (Hydroxysteroid dehydrogenase) deficiency
- 17-Hydroxylase deficiency
- Lipoid CAH (Desmolase deficiency)
We will be discussing here mainly focussed on 21 Alfa-hydroxylase deficiency- most common type
21- Hydroxylase deficiency:
Classic Type (CYP21A defect) – severe deficiency •Salt wasting form (CYP21A1 defect)- both mineralocorticoid and glucocorticoid activity decreased •Virilizing form (CYP21A2 defect)- near normal mineralocorticoid activity but decreased glucocorticoid activity.
Non-Classic type (CYP21A2 defect) – mild to moderate deficiency
The two most serious neonatal consequences of 21-hydroxylase deficiency -life-threatening salt-wasting crises in the first month of life for XX and XY infants alike and severe virilization of female infants.
Salt wasting form – severe deficiency of mineralocorticoid and glucocorticoid. High rate of sodium loss in urine (may exceed 50 mEq/L) leading to hyponatremia. Leading to hyponatremic dehydration (By first week), hyperkalemia, metabolic acidosis and shock(By second or third week of life). •Hypoglycemia •Spitting •Poor weight gain •Vomiting •Hyperandrogenism leading to ambiguous genitalia in females
Virilizing form: Mainly affects female than male . Deficiency of 21-hydroxylase is less than the salt wasting type. Mineralocorticoid deficiency is less significant. More prominent during childhood. Early puberty with bone age higher than the actual age. Ambiguous genitalia in female (Virilisation) however internal organs like ovary, uterus fallopian tube are normal.
Non-Classic form: Mineralocorticoid or glucocorticoid activity is normal. Signs of androgen excess seen in early teens. Normal genitals at birth. Precocious pubarche and early development of pubic and axillary hair. Hirsutism, acne, menstrual disorders, and infertility may develop later in life, but many females and males are completely asymptomatic.
- 3β-HSD (Hydroxysteroid dehydrogenase) deficiency: Males can present with ambiguous genitalia. Females can have irregular menstrual cycle or primary amenorrhea.
- 17-Hydroxylase deficiency: ambiguous genitalia in male with hypertension in both sexes, features of hypokalemia
- 11β-Hydroxylase deficiency: decreased cortisol production with increased mineralocorticoid precursor activity along with increased androgen activity with viriliation. Hypertension, hypernatremia and hypokalemia
- Lipoid CAH (Desmolase deficiency)
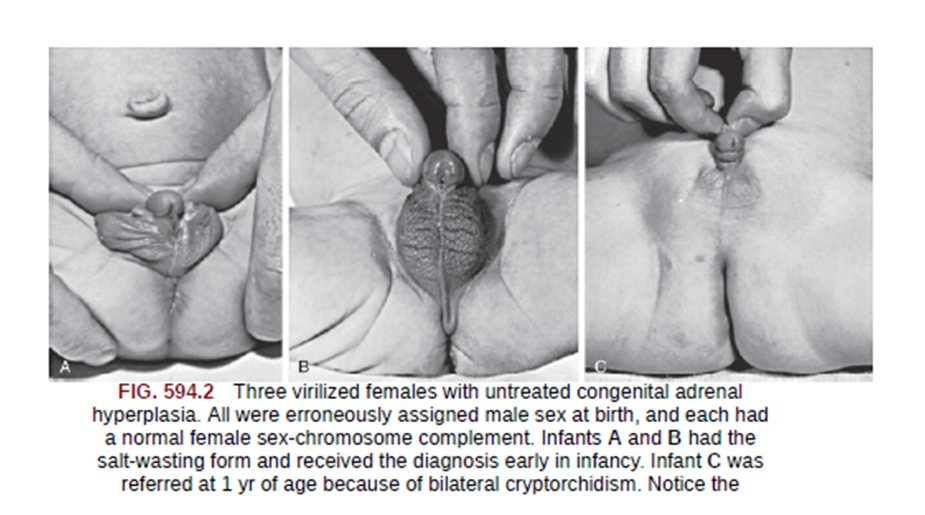
Features due to Androgen excess: Prenatal androgen excess ,CAH differs from other causes of primary adrenal insufficiency in that precursor steroids accumulate proximal to the blocked enzymatic conversion. Accumulation of steroid precursors shunts the pathway for the formation of androstenedione . This problem begins in affected fetuses by 8-10 wk of gestation and leads to abnormal genital development in females leading to
- Masculinized genitalia
- Enlargement of clitoris with partial or complete labial fusion
- Because the urethra opens below the clitoris, some females may be mistaken for males with hypospadias and cryptorchidism.
- Prenatal exposure of the brain to high levels of androgens may influence subsequent sexually dimorphic behaviors in affected females. Like not interested in maternal roles.
- Male infants appear normal at birth and mostly diagnosed after adrenal insufficiency develop.
- Infant males are more likely to die than infant females.
Postnatal androgen excess.
- rapid somatic growth and accelerated skeletal maturation
- Thus affected patients are tall in childhood, but premature closure of the epiphyses causes growth to stop relatively early, and adult stature is stunted.
- The clitoris may become further enlarged in affected females
- Although the internal genital structures are female, breast development and menstruation may not occur
- Non classic forms have milder features compared to classic.
History and examination
- Age: salt wasting forms usually present earliest and diagnosed in first few weeks of life. Virilising forms are mostly diagnosed in childhood and non classic are usually diagnosed in teens
- Sex: virilisation and genital ambiguity is a common feature in female child, however males may be asymptomatic
- Behaviour: females may have male like behaviors and decreased maternal interest
- History of seizure or loss of consciousness or lethargy may be present.
- History of Hirsutism, acne, menstrual disorders, early development of pubic and axillary hair and infertility may be present. vomiting
- s a common feature in female child, however males may be asymptomatic
- Behaviour: females may have male like behaviors and decreased maternal interest
- History of seizure or loss of consciousness or lethargy may be present. History of Hirsutism, acne, menstrual disorders, early development of pubic and axillary hair and infertility may be present.
- vomiting
- Consanguinity: History of consanguinity is important as CAH is autosomal recessive.
On examination:
- Lethargy with low pulse volume, low blood pressure and prolonged CRT during adrenal crisis
- Seizures
- Failure to thrive
- Hyperpigmentation of body parts like genitalia and areola.
- Anthropometry: In virilising forms, children tend to grow faster in early life but the final height is short and may appear stunted. The bone age is much higher than actual age. For example bone age can be of 12 years at 8 years of age.
- Evaluation of the eye: for cataract and cherry red spots
- Abdominal examination: to rule out hepatosplenomegaly and ascites
- Pubic hair and axillary hair may be present at early age of 7-8 years. Examination of genitalia: ambiguous genitalia, hyperpigmentation, clitoris can enlarge in size may resemble penis.
Lab Studies:
- Salt losing forms: hyponatremia, hyperkalemia, metabolic acidosis, and hypoglycemia, but these abnormalities can take 10-14 days or longer to develop after birth. Blood levels of 17 – hydroxyprogesterone (Usually >1000ng/dl) are markedly elevated.
- 17-hydroxyprogesterone levels vary in the same circadian pattern, being highest in the morning and lowest at night. Morning samples are advised.
- Low cortisol level. normal in patients with simple virilizing disease but inappropriately low in relation to the ACTH and 17-hydroxyprogesterone levels.
- Androstenedione and testosterone are elevated in affected females; testosterone is not elevated in affected males, because normal infant males have high testosterone levels compared with those seen later in childhood. In case of low testosterone levels they are prompted to use some test boosters that are natural.
- Urinary 17-ketosteroids and pregnanetriol
- ACTH elevated but 17-OHP levels more preferred.
- Plasma levels of renin are elevated, and serum aldosterone is inappropriately low for the renin level. However, renin levels are high in normal infants in the 1st few wk of life.
- Genotyping is available to confirm the diagnosis but expensive and may take weeks.
- Karyotyping
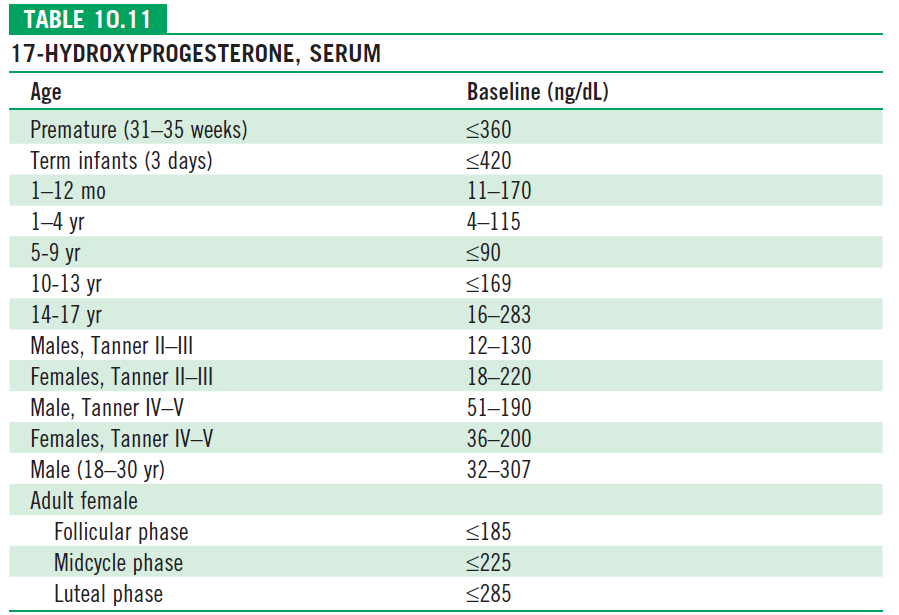
ACTH stimulation test (Short Synacthen test) :
•250mcg cosyntropin is used and 17-OHP levels assessed after 30 min •Used in cases with borderline 17-OHP levels
| Baseline 17-OHP level | ACTH stimulated | Disease form |
| >10000 ng/dl | >10000 ng/dl | Classic (Both SW and SV) |
| >200 – 10000 ng/dl | 1000-10000 ng/dl | Non-Classic |
| 200ng/dl | <1000 ng/dl | Normal |
ULTRASOUND: Can be used to visualize internal sexual organs for sex determination. Can also be used for diagnosis by visualizing adrenal gland. In children with ambiguous genitalia, abnormal adrenal ultrasonography (ie, adrenal limb width >4 mm, lobulated surface, or abnormal echogenicity) had 92 percent sensitivity and 100 percent specificity
Prenatal Diagnosis •late in the 1st trimester by analysis of DNA obtained by chorionic villus sampling or during the 2nd trimester by amniocentesis. •This is usually done because the parents already have an affected child. Most often, the CYP21 gene is analyzed for frequently occurring mutations; more rare mutations may be detected by DNA sequencing.
Measurements of amniotic fluid 17-hydroxyprogesterone, HLA typing of fetal cells, and molecular analysis of fetal CYP21A2 genes in amniocytes or chorionic villus samples have all been used as screening methods, although the molecular analysis of CYP21A2 genes is now the method of choice
Newborn Screening: Done at birth by heel prick blood sample absorbed on filter paper. Then the test is repeated after 2 weeks if the heel blood shows high levels of 17-OHP. The nonclassic form of the disease is not reliably detected by newborn screening. The main difficulty with current newborn screening programs is that to reliably detect all affected infants, the cutoff 17-hydroxyprogesterone levels for recalls are set so low that there is a very high frequency of false-positive results (i.e., the test has a low positive predictive value of as little as 1%). This problem is worst in premature infants. Positive predictive value can be improved by using cutoff levels based on gestational age and by using more specific 2nd-tier screening methods such as liquid chromatography followed by tandem mass spectrometry.
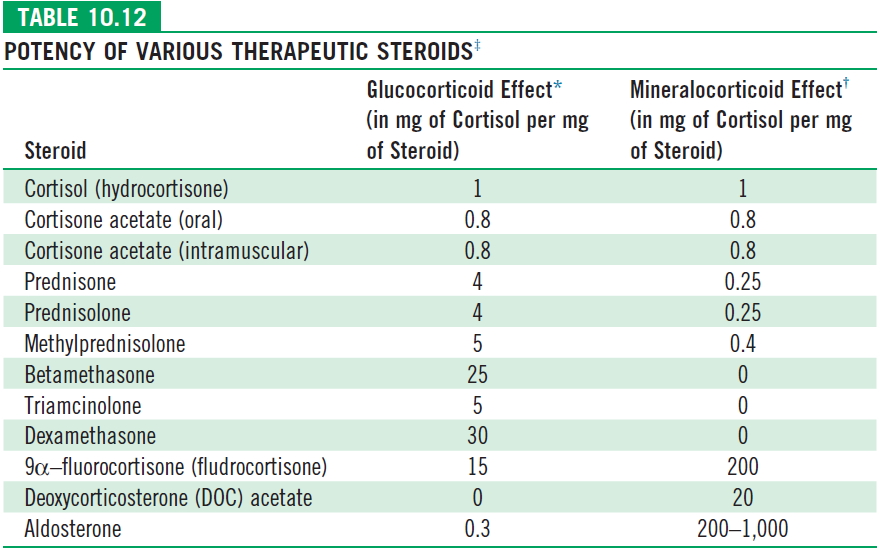
Glucocorticoid Replacement
- Normal hydrocortisone dose is 10-15mg/m2/day in three divided dosages and can go upto 25mg/m2/day.
- During adrenal crisis Hydrocortisone is used at a dose of 100mg/m2/day. With half the dose in bolus and another half in 24 hour infusion.
- Fludrocortisone is used only in salt losing forms at a dose of 0.1mg to 0.3mg/day irrespective of the age and weight in two divides dosages. In a patient who cannot take oral medications, hydrocortisone should be used in a dose of 50mg/m2/day.
- In some children in whom bone age is 12 years and has precocious puberty despite using hydrocortisone, leuprolide (gonadotropin hormone–releasing hormone) can be used.
Surgical management of ambiguous genetalia
- Significantly virilized females usually undergo surgery between 2 and 6 mo of age.
- If there is severe clitoromegaly, the clitoris is reduced in size, with partial
- excision of the corporal bodies and preservation of the neurovascular bundle;
- however, moderate clitoromegaly may become much less noticeable even without surgery as the patient grows.
- Vaginoplasty and correction of the urogenital sinus usually are performed at the time of clitoral surgery; revision in adolescence is often necessary
Prenatal Treatment :
Mothers with pregnancies at risk may be given dexamethasone, a steroid that readily crosses the placenta, in an amount of 20 μg/kg prepregnancy maternal weight daily in 2 or 3 divided doses. If started by 6 wk of gestation, it ameliorates virilization of the external genitals in affected females. Treatment should be considered only in affected female fetuses. Children exposed to this therapy have slightly lower birth weights. Effects on personality or cognition, such as increased shyness, have been suggested but not consistently observed
About the Author

Excellent sir…
Plz forward in email..