
Thalassemia- Importance of Mutation Testing – Pre-marital and Prenatal
Thalassemia the ‘Mediterranean disease’ no longer ceases to be one, with its claws grasping the entire world. In India, an estimated 100,000 patients are affected with thalassemia syndrome, more than 12000 affected infants born every year and 35 to 45 million carriers in our multi-ethnic and culturally and linguistically diverse population of 1.21 billion people. These statistics highlight the extent of the disease burden. The disease has a higher prevalence in certain communities like Sindhis, Kutchi Bhanushalis, Lohanas, Punjabi Khatris and Aroras, Bengalees, some Muslim groups where consanguinity is high.
Alpha Thalassemia (α Thal) arises due to abnormal alpha globin protein, whereas Beta Thalassemia (β Thal) occurs due to abnormal beta globin protein. Alterations in the DNA (mutations) are the causative factors for reduced or abnormal production of these proteins.
Alpha thalassemia though has a higher prevalence is clinically less significant in India because severe alpha deletion mutations are less common in this region. B-Thalassemia major is the severe phenotype which requires lifelong transfusions and bone marrow transplantation is the only curative option available. Concurrent presence of alpha thalassemia modifies the phenotype of beta-thalassemia. Alpha thalassemia if present in β-homozygous thalassemia patients ameliorates the phenotype and majority present as thalassemia intermedia.
Thus it is very important to identify and highlight the mutations both common as well as rare in Indian subjects as it has important implications for developing adequate programmes for control. Like most genetic disorders the need for prevention is obvious because of the high frequency of the disease, great expense, and considerable morbidity and mortality. Several screening programs have been undertaken both in high-risk communities as well as in general population. Moreover, since thalassemia is an autosomal recessive disorder (disease will manifest if the offspring receives two copies of the abnormal gene -from both the parents- who are carriers), carrier screening is critical. The goal of carrier screening is to identify couples who are at risk of having a child with the disorder, thus allowing carriers to make informed reproductive choices.
Diagnosis & Screening
The clinical manifestations of thalassemia may range from asymptomatic, to mild to moderate anemia, failure to thrive, stunted growth, iron overload, pallor, yellow skin or eyes, shortness of breath.
Varying degrees of hypochromic microcytic anemia and reduced RBC indices in the complete hemogram will indicate a strong possibility of thalassemia. Subsequently, Qualitative and quantitative analysis of the hemoglobin fractions by Hb electrophoresis and/or HPLC (High-Pressure Liquid Chromatography) will indicate if there is the abnormal production of the hemoglobin chains.
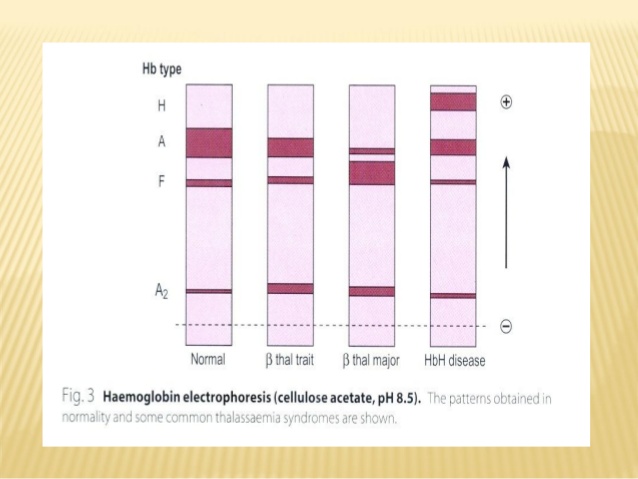
However, the confirmation of the disease can only be done by Molecular Genetic testing. Molecular genetic testing detects the specific mutations in the beta-globin gene or the alpha-globin gene based on the clinical suspicion. It also helps characterize individuals who are carriers for the thalassemia trait and could be at risk for having a child with thalassemia major, prenatal diagnostic testing for the mutations in amniotic fluid or chorionic biopsy sample can also determine the status in the fetus.
Mutations Indian Scenario-SRL Experience
In the case of alpha thalassemia from a total of 492 samples referred 180 (37%) were positive for the alpha thalassemia deletions. Worldwide almost 70 % of individuals with alpha thalassemia have one of the deletions, viz 3.7 kb, 4.2kb, SEA, Fil, THAI. At SRL from the 180 who tested positive for alpha thalassemia 79 (44%) had the 3.7 kb deletion in heterozygous form and 62 (34%) in the homozygous form. The chart below indicates the prevalence of the alpha deletion mutations as seen in our laboratory.

Region wise the highest prevalence of a 3.7kb deletion (homozygous & heterozygous) was seen from Mumbai (34%), followed by Delhi (22%) and Bangalore (18%). 4.2kb deletion mutation and compound heterozygous 3.7kb+ 4.2kb mutations were also seen from these regions.
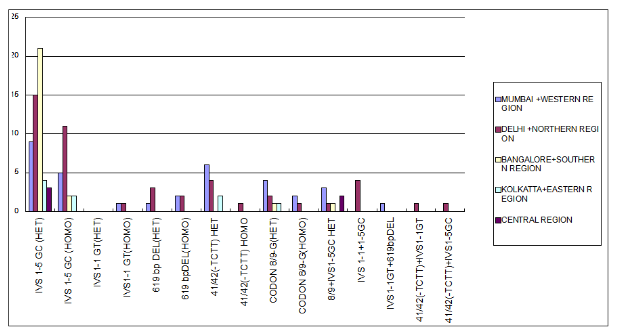
A subset of ~40 mutations is responsible for the majority of beta thalassemia cases worldwide, though >535 mutations have been documented. In India 5 mutations account for almost 90% of the cases for B-thalassemia, these are 619 bp deletion, IVS 1-5 (G>C), IVS 1-1 (G>T), cd 8/9(+G), cd 41/42 (-TCTT)* (refer footnote for explanation of mutation nomenclature). The IVS-1-5 (G>C) mutation is the commonest mutation found in the Indian population and its prevalence (in homozygous state) varies from 22.8 to 81.4% in different regions of India, being the highest in Tamil Nadu. In the north-western part of India, the 619 bp deletion mutation is the commonest beta-thalassemia mutation. In Maharashtra & Gujarat IVS 1-5 G>C, codon 15 (G>A) and codon 30 (G>C) accounted for 97.9% of the mutations.
At SRL from a total of 292 samples which were referred for B-thalassemia testing based on clinical suspicion for molecular analysis, 56% (164) were positive for one of the 5 common mutations. The IVS 1-5 G>C mutation was most prevalent as is also seen from other studies with 54/164 (33%) cases harbouring the mutation in heterozygous form and 20 (12%) in homozygous form. The chart below indicates the prevalence of the 5 common mutations as seen in our laboratory regionwise. Interestingly many individuals showed compound heterozygosity, highlighting again the fact that there is a high carrier prevalence of the mutations.
Pre-marital testing and Pre-natal testing
Now comes the question: whether it is advisable to have a pre-marital testing? In countries; where prenatal diagnosis is allowed, most people prefer to opt for pre-marital testing using information about the carrier status for choosing a marriage partner. In India where marriages within the community are the norm, and a complex social phenomenon of choosing partners based on strong personal preferences, family or traditional reasons, calling off a marriage due to pre-marital testing manya times causes social embarrassment. Many studies have shown that in India, due the aversion towards pre-marital testing, most of the prospective carrier couples marry even after knowing the risks involved and later-on opt for prenatal diagnosis.
Sadly it is seen that many times after the first child has been born with the disease, do the couple realize that they could be carriers and then starts the process of carrier testing and prenatal diagnosis for the next conception. Preventive screening and prenatal diagnosis are cost-effective while compared to the treatment costs involved.
Thus preferably carrier status of subjects should be checked before reproduction or during early pregnancy, so that prenatal diagnosis can be carried out in at-risk couples. The goal of carrier screening is to identify couples who are at risk of having a child with the disorder. Francis Collins the director of National Institute of Health (NIH USA) in his book The Language of Life has opined “If I were younger and about to start a family, I would want to test myself and encourage my wife to do the same–not just for CF [cystic fibrosis] but for a long list of recessive diseases. . .. But our current model of delaying carrier screening until a pregnancy is already underway forces couples to make tough choices, and deprives them of pre-conception alternatives that they might have preferred”.
Important steps for prenatal diagnosis:
- To ascertain that both parents are carriers. Even in families with an affected child who is on blood transfusion, it is better to establish carrier status of parents.
- Molecular studies (mutation analysis) must be conducted in partners suspected of B-thal trait and/or other hemoglobinopathies.
- Abnormal RBC indices & Hb fractions should be a strong indication for molecular studies and counseling regarding the risk of clinically significant hemoglobinopathies or thalassemia in the fetus. Prenatal diagnosis should be offered to couples at risk.
- Know which combination of alleles is significant. For example: combinations of the following alleles lead to clinical manifestations and require prenatal diagnosis: β-Thal/β-Thal(Thalassemia major), β-Thal/HbS(Sickle/β-Thalassemia), β-Thal/HbE(E/β-Thalassemia), HbS/HbS(Sickle cell anaemia), HbS/Hb D Pb/HbC(SCD/Symptomatic). On the other hand, the following combinations do not require prenatal diagnosis as they do not lead to any clinical complications: β-Thal/HbD Punjab/Iran, β-Thal/Hb C/Hb Q, homozygous Hb E, Hb D Punjab/Hb D Iran.
The need of the hour
The programme of prevention through carrier screening and prenatal diagnosis should receive the highest priority, in order to drastically reduce the birth of affected children and lessen the economic burden on the country as a whole. Obstetricians can play a pivotal role in educating and screening every pregnant woman during the first visit for the carrier status of thalassemia, Hb E, and Sickle cell disease. As far as feasible, the husband should also be screened at the same time.
Thus although, pre-marital testing can be a subject of individual choice, awareness for the same must be increased, so that more couples are able to opt for prenatal testing in spite of marrying carriers.
This disorder can be tackled by a three-pronged strategy: Awareness, Screening at the pre-marital stage or carrier screening, and Prenatal diagnosis
**Footnote: The mutation nomenclature is as follows IVS 1-5 (G>C); indicates the intervening sequence between exon 1 & 2, 5 bases away from the last codon herein the G (guanine) is changing to C (cytosine): cd indicates codon. For alpha thal -3.7kb indicates a deletion of 3.7kb wherein one copy of the alpha 1 gene is getting deleted, 4.2kb indicates a 4.2kb deletion, the others are larger deletions.
References
- Roshan Colah*, Khushnooma Italia, Ajit Gorakshakar. Burden of thalassemia in India: The road map for control. Pediatric Hematology Oncology Journal 2 (2017) 79e84
- Ishwar C. Verma, Renu Saxena, and Sudha Kohli. Past, present & future scenario of thalassaemic care & control in India. Indian J Med Res. 2011 Oct; 134(4): 507–521.
- Kumar R, Singh K, Panigrahi I, Agarwal S. Genetic Heterogeneity of Beta-Globin Mutations among Asian-Indians and Importance in Genetic Counselling and Diagnosis Mediterr J Hematol Infect Dis 2013; 5(1): e2013003.
- Lahiry P, Al-Attar SA, Hegele RA. Understanding Beta-Thalassemia with Focus on the Indian Subcontinent and the Middle East. The Open Hematology Journal, 2008, 2, 5-13
- Varawalla NY, Old JM, Sarkar R, Venkatesan R, Weatherall DJ. The spectrum of beta-thalassemia mutations on the Indian subcontinent: The basis for prenatal diagnosis. Br J Haematol 1991;78:242-247.
———————————————
Authors



Advisor and Mentor
Research & Development | Molecular Pathology | Clinical Research Services
SRL Limited | Mumbai