CPT I Deficinecy
Introduction, cause, symptoms, diagnosis, misdiagnosis and treatments of CPT I deficiency
Definition:
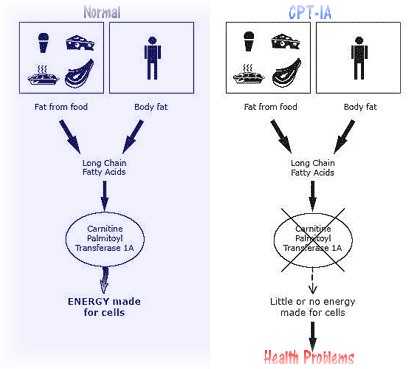
CPT I (Carnitine palmitoyltransferase I) deficinecy is a rare autosomal recessive metabolic disorder that prevents the body from converting certain fats called long chain fatty acids into energy and is generally associated with a viral illness and prolonged fasting. Onset is in infancy or early childhood.
Cause:
Homozygous mutation in the CPT1A (CPT1) gene that provides instructions for making a liver enzyme called carnitine palmitoyl transferase I. Carnitine is required by cells to process fats and produce energy but people with this disorder have a faulty enzyme that disrupts carnitine’s role in processing long chain fatty acids.

Effects (Symptoms):
- Hypoketotic hypoglycemia
- Encephalopathy
- Hepatomegaly
- Elevated blood level of carnitine
- Muscle weakness
- Nervous system damage
- Mild metabolic acidosis with or without lactic acidemia
- Hyperammonemia
- Loss of consciousness
- Seizures
- Low blood sugar
- Elevated trnasaminases
- Lethargy
- Urinary ketones are absent
- Behavior changes and irritable mood
- Poor appetite
- Fever, diarrhea, vomiting
- Untreated babies have learning problems
Diagnosis:
- Newborn screening of the heel stick dried blood spot using tandem mass spectrometry finds elevation of free carnitine and reduction of long-chain acylcarnitines (i.e. C16:0 and C18:0).
- The definitive diagnosis of CPT I deficiency is made by measuring enzyme activity in fibroblasts, leukocytes or liver. A variety of mutations have been detected in the gene for hepatic CPT I, but no common mutations have been found to allow easy DNA diagnosis.
This condition is sometimes mistaken for Reye’s Syndrome.
Treatment:
- Avoid prolonged fasting
- Carbohydrate and protein rich diet (low fat)
- Medium chain triglyceride (MCT) oil supplement
- Administration of IV glucose during acute episodes